Врожденная гиперплазия коры надпочечников
Врожденная гиперплазия коры надпочечников, которая раньше называлась адреногенитальным синдромом, является клиническим синдромом или симптомокомплексом, развитие которого связано с нарушением секреции кортикостероидов вследствие врожденного дефекта ферментов, ответственных за биосинтез этих гормонов. Сниженное образования кортизола приводит к повышению секреции АКТГ с последующим развитием гиперплазии коркового слоя коры надпочечников. Для синдрома врожденной гиперплазии коры надпочечников характерна триада: низкий уровень кортизола и высокое содержание АКТГ в крови, двусторонняя гиперплазия надпочечников. Типичным для этой патологии является интактность ренин-ангиотензин-альдостероновой системы.
Врожденная гиперплазия коры надпочечников является следствием нарушения активности ферментов, осуществляющих биосинтез стероидов. Эти ферменты контролируют гормоны не только в надпочечниках, но и в половых железах, поэтому при данной патологии имеется также нарушение секреции половых гормонов.
Врожденная гиперплазия коры надпочечников встречается не столь редко и составляет от 1:5000 до 1:67 000. Исследования показали наличие аутосомно-рецессивного пути наследования. Врожденная гиперплазия коры надпочечников может быть подразделена на следующие формы: недостаточность 21-гидроксилазы (классическая и неклассическая формы), недостаточность 11b-гидроксилазы (классическая и неклассическая формы), недостаточность 3b-гидроксистероидной дегидрогеназы, недостаточность 17a-гидроксилазы с недостаточностью 17,20-лиазы или без нее, недостаточность 20,22 десмолазы (липоидная гиперплазия коры надпочечников), недостаточность метилоксидазы I и II типа.
Недостаточность 21-гидроксилазы (Р450с21). Встречается наиболее часто из перечисленных ферментных нарушений коры надпочечников. Выраженные (классические) формы этого синдрома встречаются у новорожденных в Швеции с частотой 1:10000, а в странах северной Европы частота выявления при проведении гормональных исследований составляет от 1:500 до 1:1000. Что касается взрослой или неклассической формы этого синдрома, то частота этой патологии встречается у евреев-Ашкенази - 1:27; в Испании - 1:53: в Югославии - 1:63; в Италии - 1:333 и в гетерогенной популяции Нью-Йорка - 1:100. Таким образом, недостаточность 21-гидроксилазы, с учетом неклассической ее формы, является одним из самых часто встречающихся наследственных заболеваний. Ген Р450с21 локализуется на коротком плече 6-й хромосомы (6р) и представлен в виде активного гена (CYP21) или в виде неактивного псевдогена (CYP21P), вблизи локуса HLA, кодирующего 4-й компонент комплемента (С4А и С4В). В большинстве случаев недостаточности 21-гидроксилазы выявлены мутации CYP21, которые возникают в связи с его взаимодействием с СYP21P, что приводит к различным делециям гена, на долю которых приходится до 95% всех мутаций CYP21. При недостаточности 21-гидроксилазы блокируется конверсия 17-гидроксипрогестерона в 11-деоксикортизол, приводя к снижению образования кортизола и накоплению предшественников кортизола, т.е. 17-гидроксипрогестерона, прегненолона, 17-гидроксипрегненолона и прогестерона, которые в сетчатом слое коры надпочечников в повышенных количествах конвертируются в надпочечниковые андрогены - дегидроэпиандростерон, d4-андростендион и тестостерон. Клинически недостаточность 21-гидроксилазы протекает в виде двух форм: вирильного, сольтеряющего и неклассической формы этого синдрома.
Вирильная форма синдрома связана с частичной недостаточностью 21-гидроксилазы. Как правило, при этом наблюдается компенсация функции коры надпочечников в результате повышения секреции АКТГ, т.е. уровень кортизола в крови снижен незначительно или определяется на нижней границе нормы. Повышенная секреция АКТГ, однако, приводит к значительному образованию андрогенов, прогестерона и 17-гидроксипрогестерона, которые угнетают сользадерживающую активность альдостерона на уровне канальцев почек. Повышение уровня ренина в плазме приводит к компенсаторному усилению секреции альдостерона. Таким образом, компенсаторные механизмы (усиление секреции АКТГ), посредством которых осуществляется нормализация секреции кортизола и альдостерона, приводят к избыточному образованию андростендиона (см. "Гормоны надпочечника"). Содержание андростендиона в крови значительно повышено и хотя этот стероид обладает незначительной биологической активностью, однако на периферии он конвертируется в тестостерон, который и ответствен за развитие вирилизации. Отмечается повышение экскреции андрогенов, имеющих кислород в положении С11.Если у здоровых лиц соотношение выделения с мочой 11-дезокси-17- кетостероидов и 11-окси-17-кетостероидов составляет 4:1, то у больных вирильной формой синдрома это соотношение 1:1. Характерно также для недостаточности 21-гидроксилазы избыточное выделение прегнантриола и его метаболита 17-гидроксипрогестерона.
Клиническая картина. Вирильная форма синдрома обусловлена повышенной секрецией андрогенов, и у плода женского пола избыток приводит к маскулинизации наружных половых органов (увеличение клитора, изменение половых губ вплоть до закрытия входа во влагалище). Наружные гениталии в этих случаях приобретают вид мужских половых органов: мошонка без яичек и гипоспадия. Внутренние половые органы остаются женскими: яичники, матка с придатками. У плодов мужского пола недостаточность 21-гидроксилазы приводит к небольшим изменениям: незначительное увеличение наружных половых органов, полового члена и пигментация мошонки.
В постнатальном периоде продолжающаяся избыточная секреция андрогенов усиливает явления вирилизации. Появляются преждевременное оволосение на лобке, в подмышечных впадинах, на лице, туловище, акне. У некоторых мальчиков значительно увеличивается половой член и возникают эрекции. Отмечается ускорение роста и окостенения костей скелета, развития мышечной системы. Вначале больные обгоняют в росте своих сверстников, а в дальнейшем в связи с преждевременным закрытием зон роста отстают. У девочек также прогрессируют явления вирилизации, телосложение - по мужскому типу (рис. 29).
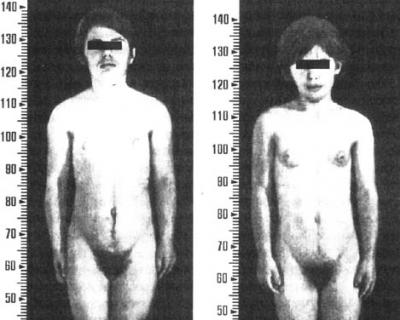
Рис 29. Больные с врожденной гиперплазией коры надпочечников.
В пубертатный период менструации не наступают, так как повышенное количество андрогенов, секретируемых надпочечниками по принципу "обратной связи", тормозит образование и выделение гонадотропинов, которое в нормальных условиях в этот период увеличивается. У мальчиков по этой же причине угнетается развитие яичек, и они остаются маленькими. Однако в некоторых случаях вирильного синдрома частично сохраняется функция половых желез, и у девочек могут наблюдаться менструации, как правило, скудные и нерегулярные, а у мальчиков - явления сперматогенеза.
Диагностика вирильной формы синдрома недостаточности 21-гидроксилазы основывается на данных клинической картины, определения экскреции с мочой 17-КС (суммарных и по фракциям) и прегнантриола, уровня андрогенов и 17-гидроксипрогестерона в плазме. Лабораторные исследования позволяют выявить повышение экскреции 11-окси-17-кетостероидов, а также андростерона, этиохоланолона, увеличение содержания в крови андростендиона и снижение уровня тестостерона. Экскреция прегнантриола с мочой резко повышена. Концентрация 17-гидроксипрогестерона в плазме крови также увеличена, тогда как содержание кортизола в крови и экскреции 17-ОКС - на нижних границах нормы. В ответ на стимуляцию АКТГ (синактен или кортросин) отмечается резкое увеличение уровня 17-гидроксипрогестерона. Этот тест используется как скрининг-тест для выявления "стертых", неклассических форм синдрома. Уровень ренина в плазме и скорость секреции альдостерона повышены.
Лечение. До применения кортикостероидов больные, как правило, при резко выраженной форме заболевания умирали в первые годы жизни при явлениях надпочечниковой недостаточности. Синтез кортикостероидов и их применение изменили судьбу этих больных. Основным видом лечения является прием кортизола или его аналогов (кортизон, преднизолон). Начальные дозы кортикостероидов должны быть в 2 раза выше физиологических, и после нормализации лабораторных показателей (17-КС и прегнантриол) дозы постепенно снижают до минимальных, при которых указанные выше показатели остаются в пределах нормы.
Новорожденные с острым надпочечниковым кризом требуют немедленной обильной гидратации с коррекцией уровня электролитов в крови и введением гидрокортизона натрий сукцината или натрий фосфата в дозе 1,5-2 мг/кг внутривенно, а затем и через рот. Для детей в возрасте до 2 лет начальные дозы составляют 30 мг кортизола (7,5 мг преднизолона), в возрасте 2 и 6 лет - соответственно 50 и 100 мг, для взрослых - 100 мг кортизола в сутки (20 мг преднизолона).
Некоторые авторы рекомендуют начинать лечение с небольших доз кортикостероидов, постепенно увеличивая их, добиваясь нормализации выделения 17-КС и прегнантриола.
Чем раньше начата терапия глюкокортикоидами, тем лучше результаты лечения. Снижение секреции АКТГ под влиянием приема глюкокортикоидов приводит к уменьшению образования андрогенов, нормализации их секреции и прекращению вирилизации организма. При различных стрессовых ситуациях (инфекция, операция, травмы и др.) дозу глюкокортикоидов необходимо увеличивать.
Сольтеряющая форма синдрома. Более глубокое нарушение, при котором имеется низкая секреция кортизола и альдостерона, несмотря на избыточное образование АКТГ. Таким образом, если при вирильной форме влияние на потерю натрия организмом избыточно образующихся предшественников кортизола (прогестерона и 17- гидроксипрогестерона) компенсируется секрецией альдостерона, то при сольтеряющей форме вследствие более глубокого нарушения дефекта 21-гидроксилазы снижено образование альдостерона и результатом такого комбинированного действия является развитие клинической картины, протекающей по типу острой недостаточности надпочечников. Уровень ренина в сыворотке крови повышен; отмечается гипертрофия юкстагломерулярного аппарата почки. Повышается содержание и ангиотензина в крови, который также способствует потере натрия через почки. Наряду с этим более резко выражены симптомы вирилизации, особенно у плодов женского пола (полное заращение половой щели и появление мошоночноподобного образования - псевдогермафродитизм).
Клиническая картина. У новорожденных при рождении выявляются указанные выше изменения наружных половых органов, пигментация кожи, снижается масса тела вследствие избыточной потери натрия и дегидратации организма. Уровень калия в крови повышен, натрия и хлоридов - снижен. На 5-10-й день после рождения развивается картина острой недостаточности надпочечников: рвота, диарея, боль в животе, апатия, и часто это состояние расценивается как пилоростеноз, для которого характерен гипокалиемической алкалоз. В отсутствие патогенетической терапии развиваются явления сердечно-сосудистой недостаточности и смерть наступает от внезапной остановки сердца вследствие гиперкалиемии.
Диагностика основывается на определении уровня электролитов в крови, а также повышенном содержании 17-гидроксипрогестерона в плазме крови и значительной экскреции с мочой прегнантриола и 17-КС. При вирилизации необходимо определение кариотипа и полового хроматина.
Лечение. Больные нуждаются в повышенном количестве жидкости и хлорида натрия. Для этой цели рекомендуется внутривенное введение раствора, содержащего 0,9% хлорида натрия и 5% глюкозы из расчета 40-60 мл на 1 кг массы тела. В некоторых случаях количество жидкости увеличивается до 100 мл/кг. При острой недостаточности надпочечников наряду с этим назначают кортизол в суточной дозе 50-75 мг. В период такой терапии необходимо опасаться развития отека легких, сердечной недостаточности и гипернатриемии. Дополнительное введение калия не показано. Как и при любом виде надпочечниковой недостаточности, противопоказаны обезболивающие вещества (морфин и др.) и барбитураты.
При выводе больного из состояния острой недостаточности надпочечников продолжают заместительную терапию, включающую наряду с приемом глюкокортикоидов (кортизола 20-25 мг в сутки, преднизолона 5-7 мг в сутки) и минералокортикоиды (ДОКА, 3-метилацетат дезоксикортикостерона, фторгидрокортизон - кортинеф или флоринеф - 0,05-0,1-0,2 мг в сутки). Адекватность заместительной терапии определяется по экскреции 17-КС и прегнантриола и нормализации секреции АКТГ.
Неклассическая форма недостаточности 21-гидроксилазы. Иногда это нарушение приобретенной или "взрослой" формы недостаточности 21-гидроксилазы. При рождении новорожденные женского пола имеют нормальные гениталии. В подростковом и пубертатном возрасте развивается гирсутизм, акне, нерегулярные менструации и даже бесплодие. Описано достаточное количество женщин, у которых после замужества при наличии нерегулярных месячных наступала беременность и роды, а в последующем наступала аменорея, бесплодие и при обследовании обнаруживались поликистозные яичники. Обследование таких женщин должно включать определение базального уровня 17-гидроксипрогестерона в крови и повторное исследование после стимуляции АКТГ. В норме содержание 17-гидроксипрогестерона через 60 минут после введения АКТГ не превышает 300-330 нг/100 мл. У больных с неклассической или поздней формой синдрома его уровень повышается до 1500 нг/100мл. Генетические исследования выявляют при этом ген HLA B14 или В47 в сочетании с CYP21B. Лечение неклассической формы недостаточности 21-гидроксилазы такое же, однако дозы глюкокортикоидов обычно ниже, чем при классических формах синдрома.
Недостаточность 11b-гидроксилазы. Нарушение биосинтеза кортикостероидов, связанное с недостаточностью 11b-гидроксилазы, встречается 1:100 000 в общей кавказоидной популяции (M. Zachmann и соавт., 1983), а в Израиле эта патология выявляется у 1:5000 до 1:7000 новорожденных (A. Rosler, 1992). Недостаточность 11b-гидроксилазы составляет около 5% от общего количества больных с врожденной гиперплазией коры надпочечников. 11b-гидроксилаза необходима для нормального синтеза глюкокортикоидов и минералокортикоидов. Имеется две изоформы этого фермента, которые кодируются двумя различными генами, локализованными на 8q21-q22 (M.J. Wagner и соавт., 1991). Основным считается ген CYP11B1 (или Р450XIB1), который экспрессируется в нормальных надпочечниках, и его транскрипция регулируется АКТГ посредством его вторичного мессенджера цАМФ. Второй ген CYP11B2 также экспрессируется в надпочечниках, но его транскрипция осуществляется на очень низком уровне. Последняя изоформа фермента называется также Р450XIB2 (или Р450aldo) и осуществляет гидроксилирование в 11-м положении, переводя 11- дезоксикортикостерон (DOC) в кортикостерон и 11-деоксикортизол в кортизол. В результате этой реакции (гидроксилирования в 18-м положении) через стадию 18-гидроксикортикостерона образуется альдостерон. Белок гена CYP11B1 (или Р450XIB1) осуществляет в основном гидроксилирование в 11-м положении, хотя и незначительно, но гидроксилирует углерод и 18-м положении. Считается, что Р450XIB1 локализуется в пучковой зоне, где контролирует синтез кортизола, а Р450XIB2 - в клубочковой зоне, где осуществляет синтез альдостерона. Показано, что мутация гена, функционирующего в пучковой зоне, приводит к нарушению синтеза кортизола и накоплению дезоксикортикостерона и его предшественников, вызывающих гипертензию. В случае мутации гена, функционирующего в клубочковой зоне, происходит нарушение синтеза альдостерона, что сопровождается клинической картиной сольтеряющего синдрома. У большинства больных недостаточность 11b-гидроксилазы приводит к повышенному образованию 11-дезоксикортикостерона, который, как известно, оказывает минералокортикоидное действие, способствуя задержке натрия в организме и развитию артериальной гипертензии. Гипертензия может быть незначительной или даже отсутствовать у детей раннего возраста. Недостаточное образование кортизола приводит по принципу "обратной связи" к гиперсекреции АКТГ, гиперплазии надпочечников, и наряду с повышением образования дезоксикортикостерона наблюдается увеличение секреции андрогенов, способствующих развитию различной степени вирилизации. Гипертензия, как правило, не коррелирует с наличием и степенью выраженности гипокалиемии или со со степенью выраженности вирилизации. При этой патологии отсутствует гиперплазия юкстагломерулярного аппарата почки и уровень ренина в плазме крови не повышен.
Помимо классической формы недостаточности 11b-гидроксилазы, описаны ее неклассические формы. Так, M.D. Birnbaum и L.I. Rose (1984) установили, что недостаточность 11b-гидроксилазы в зрелом возрасте сочетается с нарушениями менструальной функции.
Диагностика недостаточности 11b-гидроксилазы основывается на клинической картине (артериальная гипертензия и вирилизация). Если у больных с недостаточностью 21-гидроксилазы из-за недостаточности альдостерона нарушается способность к сохранению натрия в организме, то у больных с недостаточностью 11b-гидроксилазы вследствие избытка дезоксикортикостерона и других агонистов ДОКа имеют место задержка натрия в организме, увеличение объема циркулирующей жидкости, гипокалиемия, снижение рениновой активности в плазме и гипертензия. При лабораторном исследования выявляются увеличение уровня 11-деоксикортизола и дезоксикортикостерона в сыворотке крови и повышение экскреции их тетрагидропроизводных (повышение экскреции 17-КС, прегнантриола, тетрагидрокортизола и его производных). Отмечается резкое снижение или отсутствие 11-окисленных С19 и С21 стероидов в крови и моче. При пробе со стимуляцией АКТГ имеет место характерное увеличение 11-деоксикортизола и ДОКа. Проба с дексаметазоном приводит к снижению выделения 17-КС и 17-гидроксикортикостероидов (дериваты тетрагидрокортизона).
Заместительная терапия глюкокортикоидами приводит к нормализации АКТГ, дезоксикортикостерона и андрогенов, что клинически проявляется нормализацией артериального давления и уменьшением или исчезновением симптомов вирилизации.
Недостаточность 3b-гидроксистероидной дегидрогеназы. Сравнительно редкая форма врожденной гиперплазии коры надпочечников, которая была описана в 1962 г. A.M. Bongiovanni как адреногенитальный синдром, связанный с недостаточностью 3b-гидроксистероидной дегидрогеназы. Этот фермент необходим для синтеза биологически активных d4-надпочечниковых стероидов и половых гормонов (d4-3-оксо/кето/-стероиды) из относительно биологически неактивных предшественников - d5-стероидов (3 b-гидрокси-d5-стероидов). Недостаточность трех b-гидроксистероидных дегидрогеназ/d5-d4-изомераз (или 3b-гидроксистероидной дегидрогеназы) является, таким образом, одной из форм врожденной гиперплазии надпочечников. Установлено, что у человека контроль синтеза фермента осуществляется двумя генами, ответственными за синтез: ген 3b-гидроксистероидной дегидрогенезы I типа, который специфически экспрессируется в плаценте и различных периферических тканях (кожа, молочная железа, печень) и ген 3b-гидроксистероидная дегидрогеназа II типа, который преимущественно экспрессируется в надпочечниках и половых железах. Недостаточность 3b-гидроксистероидной дегидрогеназы связана с мутацией соответствующего гена. R. Sanchez и соавт. (1989,1991,1994) охарактеризовали клоны кДНК, кодирующие 3b-гидроксистероидную дегидрогеназу I и II типа. Затем было показано, что оба гена состоят из 4 экзонов и 3 интронов. Локус 3b-гидроксистероидной дегидрогеназы локализуется на 1-й хромосоме (1р13). Показано, что недостаточность 3b-гидроксистероидной дегидрогеназы обусловлена мутацией гена 3b-гидроксистероидной дегидрогеназы II типа (M. Zerah и соавт., 1994). 3b-Гидроксистероидная дегидрогеназа является необходимой для биосинтеза всех классов стероидов: прогестины, минералокортикоиды, глюкокортикоиды, андрогены и эстрогены. Поэтому недостаточность этого фермента проявляется нарушением образования кортизола, альдостерона, а также снижением конверсии дегидроэпиандростерона в андростендион и последнего в тестостерон. В этой связи в сыворотке крови таких больных избыточно определяется прегненолон, 17a-гидроксипрегненолон и дегидроэпиандростендион. 3b-Гидроксистероидная дегидрогеназа необходима также для образования половых гормонов в гонадах, что ведет к недостаточному образованию андростендиона и тестостерона.
У новорожденных обоих полов при недостаточности 3b-гидроксистероидной дегидрогеназы выявляется клиническая картина сольтеряющего синдрома различной степени тяжести. У новорожденных мужского пола эти нарушения сочетаются с псевдогермафродитизмом, тогда как у новорожденных женского пола имеется нормальная дифференцировка наружных половых органов и умеренная вирилизация вследствие того, что дегидроэпиандростерон оказывает слабое андрогенное действие. Если при тяжелой степени сольтеряющего синдрома болезнь не диагностируется в первые дни, то прогноз заболевания может оказаться серьезным. Предположить правильный диагноз у мальчиков позволяет неопределенность наружных половых органов и гипоспадия. Однако при неполной недостаточности фермента, даже при наличии нарушений развития наружных половых органов у мальчиков, правильный диагноз в некоторых случаях устанавливается только в пубертатном периоде. Дифференцировка наружных половых органов у девочек в норме или может иметь место только умеренная клиторомегалия. Поэтому часто диагноз устанавливается только при наступлении адренархе.
Помимо классической формы недостаточности 3b-гидроксистероидной дегидрогеназы, чаще встречается неклассическая форма заболевания, при которой вирилизация развивается после адренархе или наступления пубертата. Это аутосомно-рецессивное нарушение биосинтеза кортикостероидов, которое по данным некоторых авторов встречается даже чаще, чем неклассическая форма недостаточности 21-гидроксилазы. P. Schram и соавт. (1992) указывают, что эта форма заболевания часто расценивается как следствие избыточной секреции андрогенов (гиперандрогения) с последующим гирсутизмом, акне и бесплодием. Характерным для данной патологии является низкорослость, причем часто у девочек рост на 5-10 см ниже, чем можно предполагать исходя из роста их родителей. В 50% случаев выявляются при этом поликистозные яичники. Эти исследователи указывают, что из 700 женщин при наличии в клинической картине признаков избытка андрогенов у 16% при обследовании была выявлена неклассическая форма 3b-гидроксистероидной недостаточности. У мальчиков вирилизация проявляется в виде ускоренного роста и увеличения наружных половых органов, тогда как у девочек отмечается умеренная вирилизация вследствие того, что дегидроэпиандростерон оказывает слабое андрогенное действие.
При гормональном обследовании выявляется увеличение содержания андрогенов в сыворотке крови. Проба с АКТГ показывает, что через 60 мин после введения уровень прегненолона в сыворотке крови повышается до 1600 нг/100мл или выше; содержание дегидроэпиандростерона - до 1800 нг/мл и выше; отношение прегненолона к 17-гидроксипрогестерону 60 или выше, а отношение концентрации прегненолона к кортизола 50 или выше (T. Eldar-Geva и соавт., 1990).
Лечение проводится кортизолом или преднизолоном, прием которых приводит к снижению секреции АКТГ и нормализации секреции кортикостероидов. Чаще всего проводится терапия дексаметазоном по 0,25 или 0,5 мг на ночь. Клинический эффект оценивается через 3-4 месяца лечения. В некоторых случаях требуется назначение минералокортикоидов (фторгидрокортизона). При значительном гирсутизме дополнительно рекомендуется спиронолактон или комбинация эстрогенов с прогестероном в низких дозах. У некоторых женщин с вирилизмом и мускулинизацией положительный эффект получен от использования нестероидного антиандрогена - флютамида. У женщин при неклассической форме синдрома с наличием поликистозных яичников нормализация активности 3b-гидроксистероидной дегидрогеназы наблюдалась через 3 месяца лечения агонистами гонадолиберина.
Недостаточность 17a-гидроксилазы (Р450сa). Цитохром Р450с17 является ферментом, который обладает активностью как 17a-гидроксилазы, так и 17,20-лиазы. Установлено, что это соединение кодируется геном, который экспрессируется как в надпочечниках, так и в половых железах. Ген, называемый также CYP17, локализуется на 10-й хромосоме (q 24-25) и состоит из 8 экзонов. Недостаточность 17a-гидроксилазы приводит к частичной или полной блокаде образования кортизола, тогда как секреция кортикостерона и дезоксикортикостерона не нарушается. Недостаток секреции кортизола является причиной повышения секреции АКТГ с последующей гиперплазией коры надпочечников, где стероидогенез сдвигается в сторону избыточного образования дезоксикортикостерона, уровень которого в плазме крови значительно повышен. Под влиянием избытка дезоксикортикостерона отмечаются задержка натрия в организме и артериальная гипертензия. Наблюдающаяся при этом гиперволемия вызывает снижение активности ренин-ангиотензинной системы. Секреция альдостерона также значительно понижена. 17-Гидроксилирование является необходимым этапом в образовании андрогенов, поэтому оно также уменьшается и, естесственно, приводит к снижению образования эстрогенов. В пубертатном периоде вторичные половые признаки у девочек выражены недостаточно или отсутствуют и имеет место первичная аменорея.
В связи с тем, что кортикостерон обладает глюкококортикоидной активностью, недостаточность глюкококортикоидов не проявляется так резко, как при дефекте других ферментов стероидогенеза. Однако повышение кортикостерона и дезоксикортикостерона способствует развитию гипертонии и гипокалиемии. Недостаток андрогенов приводит к нарушению формирования наружных половых органов у мальчиков с явлениями псевдогермафродитизма, а в пубертатном возрасте - к развитию генекомастии. Отсутствие функционального ответа яичек на действие гонадотропных гормонов свидетельствует об инфантилизме у таких мальчиков и девочек в пубертатном периоде. Применение тестостерона в этот период приводит к маскулинизации. Для диагностики недостаточности 17a-гидроксилазы наряду с наличием у мальчииков псевдогермафродитизма необходимо определение полового хроматина и кариотипа, который способствует мужскому полу - XY. В крови определяется повышение содержания АКТГ, кортикостерона и дезоксикортикостерона при снижении уровня кортизола и альдостерона.
У девочек диагностика этого нарушения затруднена, так как наружные половые органы развиты нормально и лишь в период полового созревания выявляется недостаточность функции яичников. Наличие гипертонии и гипокалиемии позволяет заподозрить это заболевание и провести соответствующее определение уровня гормонов в крови. Для недостаточности 17-a-гидроксилазы установлен аутосомно-рецессивный тип наследования.
Заместительная терапия глюкокортикоидами приводит к снижению секреции АКТГ, уменьшению образования кортикостерона и дезоксикортикостерона и нормализации артериального давления. В препубертатный период показано назначение половых гормонов.
В случае задержки яичек в брюшной полости и развития наружных половых органов у мальчиков по женскому типу некоторые ученые рекомендуют удаление таких яичек из-за возможности их злокачественного перерождения; после этого проводится заместительная терапия женскими половыми гормонами.
Недостаточность кортикостерон метилоксидазы II типа. Проявляется снижением образования альдостерона, тогда как биосинтез предшественников альдостерона и дезоксикортикостерона остается ненарушенным. Клинически недостаточность кортикостерон метилоксидазы II типа протекает в виде сольтеряющего синдрома. Биосинтез других кортикостероидов (кортизола и др.) не нарушен и секреция АКТГ у таких больных в норме, поэтому отсутствуют признаки вирилизации и пигментации, характерные для этих форм врожденной гиперплазии надпочечников. В большинстве случаев недостаточность кортикостерон метилоксидазы II типа протекает в компенсированной форме; кортикостерон и дезоксикортикостерон оказывают незначительное минералокортикоидное действие и, как правило, дополнительного приема хлорида натрия с пищей достаточно для поддержания обмена электролитов на нормальном уровне. Лишь в некоторых случаях для этого требуется назначение фторкортизона или других минералокортикоидов (ДОКСА и др.).
Липоидная гиперплазия надпочечников. Наиболее тяжелое нарушение биосинтеза кортикостероидов, при котором имеется недостаточность десмолазы (20,22-десмолазы или Р450scc), осуществляющей наиболее ранний этап биосинтеза, а именно образование прегненолона из холестерина. Ген, кодирующий десмолазу (CYP11A), локализуется на 15-й хромосоме. При недостаточности этого фермента блокируется синтез альдостерона, кортизола, андрогенов, и такая патология несовместима с жизнью. Новорожденные умирают в первые дни жизни. На вскрытии обнаруживаются увеличенные в размерах надпочечники с высоким содержанием липидов, в основном холестерина, что и послужило основанием для названия этого нарушения-липоидная гиперплазия надпочечников. Нарушение биосинтеза стероидов наблюдается не только в надпочечниках, но и в половых железах, и у плодов мужского пола в отсутствие андрогенов развитие наружных половых органов происходит по женскому типу. Правильное установление пола в этих случаях возможно лишь при определении полового кариотипа. При ранней диагностике необходима немедленная заместительная терапия, включающая кортизол, минералокортикоиды, а также дополнительный прием хлорида натрия. Прогноз неблагоприятный.
Почитайте ещё медицинских статей:
- Белое и серое вещество моста Мост, pons (варолиев мост), является производным заднего мозга, metencephalon, и представляет собой почти четырехсторонней формы большой белый вал, лежащий кзади от центра основания мозга. Спереди он резко отграничен от ножек
- Мышцы верхней конечности Мышцы верхней конечности, mm. membri superioris. соответственно их топографоанатомическим особенностям разделяются на две группы: мышцы пояса верхней конечности, mm. cinguli membri superioris, и мышцы свободной верхней конечности, mm. partis liberae
- Поверхностные лимфатические сосуды К паховым лимфатическим узлам подходят также поверхностные лимфатические сосуды от наружной поверхности бедра, ягодичной области и нижних отделов спины. Выносящие лимфатические сосуды поверхностных паховых лимфатических узлов прободают широкую фасцию бедра
Размещена в категории Эндокринная система